Our research deals with quantum and semiclassical theories of chemical dynamics and thermodynamics for "real" molecular systems. Development of such theories/methods would lead to prediction of dynamic, spectroscopic, and thermodynamic properties where nuclear quantum effects (including zero point energy, tunneling, and coherence effects) become important at low temperatures and/or in realistic systems that contain light atoms such as hydrogen, helium, and even lithium. Typical examples include vibrational dynamics, ground-/excited-electronic-state proton transfer reactions, hydrogen bonding effects, H/D/T isotopic substitution, separation, and fractionation, and many other phenomena (of importance of nuclear quantum effects) in chemical, biological, and materials systems. In addition to infrared (IR) and Raman spectroscopy, elastic and inelastic neutron scattering, high-resolution scanning tunneling microscopy, and other experimental methods, developing theories and computational tools to understand these phenomena or experimental results at the atomistic level presents challenging frontiers in modern physical chemistry.
We are focused on developing new methods and models, and also on applying them to "real" molecular systems. Current primary interests are
Quantum/Classical Statistical Mechanics
Efficient thermostats for molecular dynamics and path integral molecular dynamics
Path integral molecular dynamics for nonadiabatic systems
Quantum Dynamics and Spectroscopy for Complex/Large Systems
Path integral Liouville dynamics
Semiclassical dynamics
Phase space formulations of quantum dynamics
Trajectory-based nonadiabatic quantum dynamics
Machine Learning for Quantum Dynamics and Statistical Mechanics
We have proposed a generic framework to construct mapping models in the Cartesian phase space for the multi-state Hamiltonian operator [J. Chem. Phys. 145, 204105 (2016)]. After mapping an F-dimensional Hamiltonian onto an F+1 dimensional space as suggested in Phys. Rev. Lett. 78, 578 (1997), creation and annihilation operators are defined such that the F+1 dimensional space is complete for any combined excitation. Commutation and anti-commutation relations are then naturally derived, which show that the underlying degrees of freedom are neither bosons nor fermions. The framework provides three mapping approaches: (a) from the analogy with the quantum-classical correspondence for two non-commutable operators; (b) from the analogy with the classical angular momentum; and (c) from the analogy with the classical vector. While approach (a) offers a novel means to derive the well-known Meyer-Miller Hamiltonian model [J. Chem. Phys. 70, 3214 (1979)], approaches (b) and (c) set the scene for developing other exact Hamiltonian formulations and their semiclassical/quasiclassical counterparts for multi-state dynamics/nonadiabatic dynamics.
We have further introduced the isomorphism between the multi-state Hamiltonian and the second-quantized many-electron Hamiltonian (with only 1-electron interactions) and demonstrated that all methods developed for the former can be used for the latter, and vice versa. It is shown that the recently proposed mapping model for the many-electron Hamiltonian [J. Chem. Phys. 137, 154107 (2012)] is not unique but one of a few exact mapping models naturally derived in the framework.
The relevant peer-reviewed papers are
"A unified theoretical framework for mapping models for the multi-state Hamiltonian"
Jian Liu*
Journal of Chemical Physics, 145, 204105 (2016)
"Isomorphism between the multi-state Hamiltonian and the second-quantized many-electron Hamiltonian with only 1-electron interactions"
Jian Liu*
Journal of Chemical Physics, 146, 024110 (2017)
"A new perspective for nonadiabatic dynamics with phase space mapping models"
Xin He, Jian Liu*
Journal of Chemical Physics, , 151, 024105 (2019). (Invited contribution to Special Topic Issue on "Dynamics of Open Quantum Systems")
"Negative Zero-Point-Energy Parameter in the Meyer–Miller Mapping Model for Nonadiabatic Dynamics"
Xin He, Zhihao Gong, Baihua Wu, and Jian Liu*
Journal of Physical Chemistry Letters, 12, 2496–2501 (2021)
"Commutator Matrix in Phase Space Mapping Models for Nonadiabatic Quantum Dynamics"
Xin He, Baihua Wu, Zhihao Gong, Jian Liu*
Journal of Physical Chemistry A, 125, 31, 6845–6863 (2021)
[https://doi.org/10.1021/acs.jpca.1c04429] [Invited contribution to the Tanimura Festschrift]
"Unified formulation for phase space mapping models for nonadiabatic quantum dynamics"
Jian Liu*, Xin He, Baihua Wu
Accounts of Chemical Research, 54, 23, 4215–4228 (2021)
https://doi.org/10.1021/acs.accounts.1c00511 [Invited contribution]
"New Phase Space Formulations and Quantum Dynamics Approaches"
Xin He, Baihua Wu, Youhao Shang, Bingqi Li, Xiangsong Cheng, Jian Liu*
Wiley Interdisciplinary Reviews-Computational Molecular Science, e1619 (2022)
https://doi.org/10.1002/wcms.1619 Supporting Information [Invited contribution]
"Nonadiabatic Field on Quantum Phase Space: A Century after Ehrenfest"
Baihua Wu, Xin He, Jian Liu*
The Journal of Physical Chemistry Letters, 15, 2, 644-658 (2024)
https://doi.org/10.1021/acs.jpclett.3c03385 Supporting Information [Invited Perspective]
"A Novel Class of Phase Space Representations for the Exact Population Dynamics of Two-State Quantum Systems and the Relation to Triangle Window Functions"
Xiangsong Cheng, Xin He, Jian Liu*
Chinese Journal of Chemical Physics, 37, 230-254 (2024)
https://doi.org/10.1063/1674-0068/cjcp2403033 [Editor's Pick. Invited contribution to the Special Issue "In memory of Prof. Qihe Zhu on the occasion of his 100th Anniversary"]
"Nonadiabatic Field with Triangle Window Functions on Quantum Phase Space"
Xin He, Xiangsong Cheng, Baihua Wu, Jian Liu*
Journal of Physical Chemistry Letters, 15, 5452-5466 (2024)
https://doi.org/10.1021/acs.jpclett.4c00793 Supporting Information
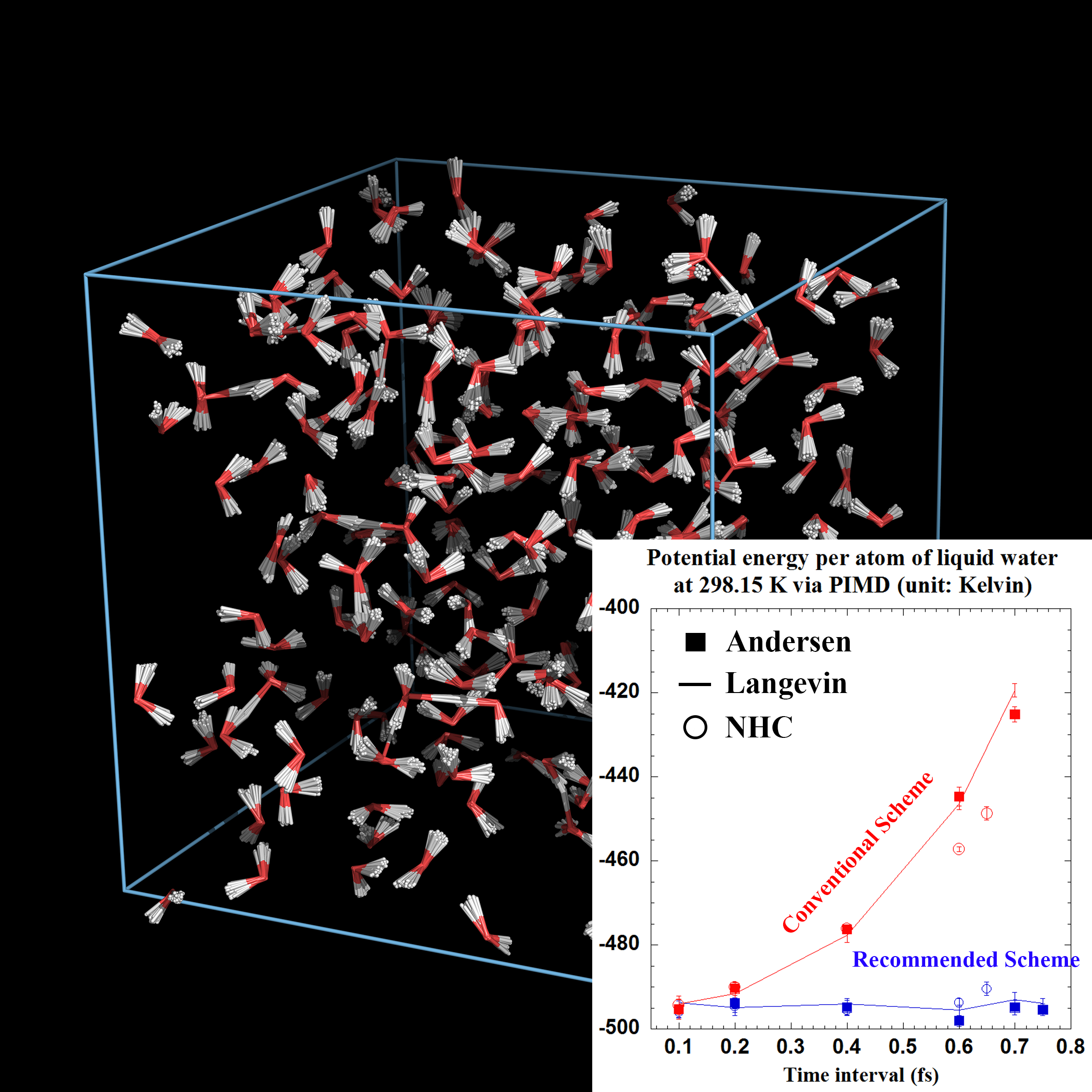
In the unified framework we have revealed that, in addition to the usual density evolution, there exists another type of discrete evolution that may not correspond to a continuous, real dynamical counterpart of the Langevin equation [J. Chem. Phys. 147, 184104 (2017)]. This virtual dynamics case is also amenable to the desired Boltzmann distribution for various stochastic thermostatting methods [Chin. J. Chem. Phys. 30, 735 (2017)]. By analyzing the asymptotic distribution and characteristic correlation time, we have shown that, for good numerical performance in efficiency as well as accuracy, one may choose the thermostat parameter (of the “middle” scheme) in a wide range from around the optimal value to the high value limit.
More recently, we have further used the unified framework to develop a novel practical PIMD methodology in either of the diabatic and adiabatic representations for studying exact quantum statistics of large multi-electronic-state systems in thermal equilibrium when the Born-Oppenheimer approximation, Condon approximation, and harmonic bath approximation are no longer valid [J. Chem. Phys. 148, 102319 (2018)].
Note: The unified "middle" thermostat scheme has been implemented in AMBER (2019 version).
The relevant peer-reviewed papers are
"A simple and accurate algorithm for path integral molecular dynamics"
Jian Liu*, Dezhang Li, Xinzijian Liu
Journal of Chemical Physics, 145, 024103 (2016)
"A unified thermostat scheme for efficient configurational sampling for classical/quantum canonical ensembles via molecular dynamics"
Zhijun Zhang, Xinzijian Liu, Zifei Chen, Haifeng Zheng, Kangyu Yan, Jian Liu*
Journal of Chemical Physics, 147, 034109 (2017)
"Path integral molecular dynamics for exact quantum statistics of multi-electronic-state systems"
Xinzijian Liu, Jian Liu*
Journal of Chemical Physics, 148, 102319 (2018)
[Invited article for the JCP special topic issue on "Nuclear Quantum Effects"]
"Stationary state distribution and efficiency analysis of the Langevin equation via real or virtual dynamics"
Dezhang Li, Xu Han, Yichen Chai, Cong Wang, Zhijun Zhang, Zifei Chen, Jian Liu*, Jiushu Shao*
Journal of Chemical Physics, 147, 184104 (2017)
"Understanding molecular dynamics with stochastic processes via real or virtual dynamics"
Dezhang Li, Zifei Chen, Zhijun Zhang, Jian Liu*
Chinese Journal of Chemical Physics, 30, 735 (2017)
[Invited article for the CJCP special topic issue on "Chemical Dynamics"]
"A unified efficient thermostat scheme for the canonical ensemble with holonomic or isokinetic constraints via molecular dynamics"
Zhijun Zhang, Xinzijian Liu, Kangyu Yan, Mark Tuckerman, Jian Liu*
Journal of Physical Chemistry A, 123, 28, 6056-6079 (2019)
[Invited contribution to "Young Scientist Virtual Special Issue"]
https://doi.org/10.1021/acs.jpca.9b02771][pdf version] (Highlighted in JPC Virtual Issue on New Tools and Methods )
"Extensive Numerical Tests of the Leapfrog Integrator in the Middle Thermostat Scheme in Molecular Simulations"
Zhaoxi Sun, Payam Kalhor, Yang Xu, Jian Liu*
Chinese Journal of Chemical Physics, 34, 6, 932-948 (2021)
[Invited contribution to the John Zhang Festschrift] https://doi.org/10.1063/1674-0068/cjcp2111242
"Generalized Fourth-order Decompositions of Imaginary Time Path Integral: Implication of the Harmonic Oscillator"
Cong Wang, Lihan Zhang, Jian Liu*, Jiushu Shao*
Chinese Journal of Chemical Physics, 35(3), 516-536 (2022)
[Invited Contribution to the Special Topic "Quantum and Classical Dynamics in Chemistry"] https://cps.scitation.org/doi/pdf/10.1063/1674-0068/cjcp2205089
We were the first to develop a practical quantum dynamics method satisfying the two critical fundamental criteria: conservation of the quantum Boltzmann distribution for the thermal equilibrium system and being exact for any thermal correlation functions (of even nonlinear operators, i.e., nonlinear functions of position or momentum operators) in the classical and harmonic limits. The so-called path integral Liouville dynamics (PILD) [J. Chem. Phys. 140, 224107 (2014)] was derived from the elaborate combination of the imaginary time path integral and the three new theoretical frameworks that we formulated for generating trajectory-based dynamics methods in the quantum phase space [J. Chem. Phys. 134, 104101 (2011); 134, 104102 (2011); 134, 194110 (2011). Scientia Sinica Chimica, 46, 27 (2016)].
PILD was applied to vibrational dynamics of several simple but representative realistic molecular systems (OH, water, ammonia, and methane) [J. Chem. Phys. 144, 034307 (2016)]. The dipole-derivative autocorrelation function was employed to obtain the infrared spectrum as a function of temperature and isotopic substitution. Comparison to the exact vibrational frequency shows that PILD produces a reasonably accurate peak position with a relatively small full width at half maximum. PILD offers a potentially useful trajectory-based quantum dynamics approach to compute vibrational spectra of molecular systems. We are extending the approach to study larger systems.
The relevant peer-reviewed papers are
“Path integral Liouville dynamics for thermal equilibrium systems”
Jian Liu*
Journal of Chemical Physics, 140, 224107 (2014)
"Further study of path integral Liouville dynamics"
Jian Liu*, Dezhang Li, Xinzijian Liu
Scientia Sinica Chimica, 46, 27 (2016)
[Invited article for the special issue for the Festschrift for Professor Lemin Li's 80th birthday]
"Path integral Liouville dynamics: Applications to infrared spectra of OH, water, ammonia, and methane"
Jian Liu*, Zhijun Zhang
Journal of Chemical Physics, 144, 034307 (2016)
Zhijun Zhang, Zifei Chen, Jian Liu*
"Path integral Liouville dynamics simulations of vibrational spectra of formaldehyde, and hydrogen peroxide"
Chinese Journal of Chemical Physics, 33, 613 (2020)
[https://doi.org/10.1063/1674-0068/cjcp2006099] [Invited contribution]
Xinzijian Liu, Linfeng Zhang, Jian Liu*
"Machine Learning phase space quantum dynamics approaches"
Journal of Chemical Physics, 154, 184104 (2021)
[https://doi.org/10.1063/5.0046689]
[Invited contribution to the Special Topic 'Quantum Dynamics with ab Initio Potentials']
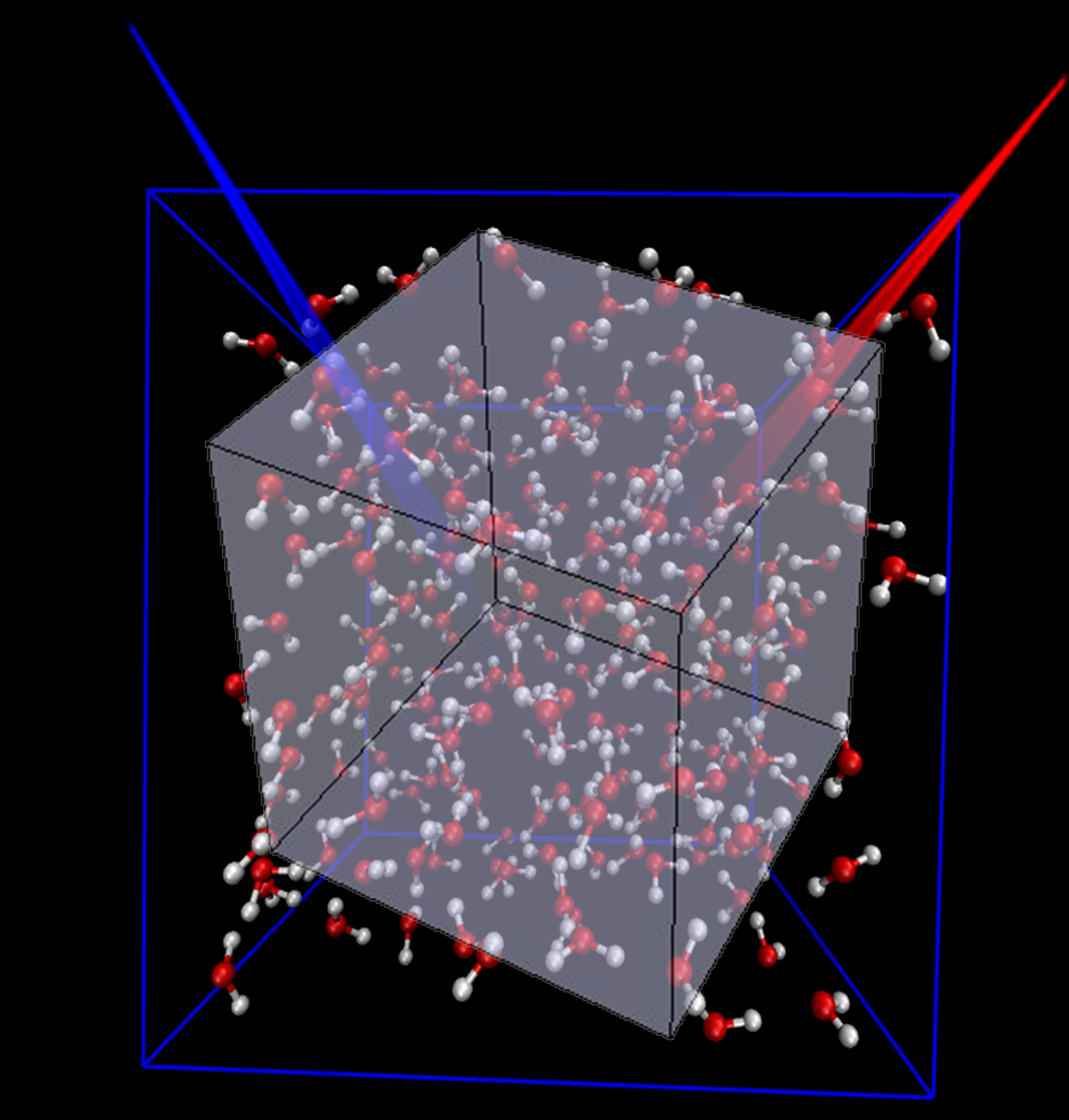
Understanding vibrational spectra (infrared (IR) and Raman spectra) at the atomistic level is important for the elucidation of dynamical processes in complex/large molecular systems. The correlation approach offers a bridge to connect the microscopic motion to the experimental spectrum. Because the dipole moment/polarizability (or its time derivative) is often a highly nonlinear function of coordinates or/and momenta, we have employed the linearized semiclassical initial value representation for quantum dynamical simulations of liquid water (and heavy water) under ambient conditions based on an ab initio based, flexible, polarizable model. Besides applying our new PIMD algorithms in the “middle” scheme, we have proposed a new technique (by using the finite difference along the direction of a related vector) to significantly reduce the computational cost for preparing the initial condition for the correlation function.
It is shown that quantum dynamical effects play a critical role in reproducing the peaks in the intermediate region between the librational and bending bands, those between the bending and stretching bands, and the double-peak in the stretching band in the experimental isotropic Raman spectrum. In contrast, quantum dynamical effects are important but less decisive in the anisotropic Raman spectrum. By selectively freezing either the intramolecular O-H stretching or H-O-H bending mode, we demonstrate that the peak in the intermediate region (2000-2400 cm-1) of the isotropic Raman spectrum arises from the interplay of the stretching and bending motions while a substantial part of the peak in the same intermediate region of the anisotropic Raman spectrum may be attributed to the combined motion of the bending and librational modes.
The relevant peer-reviewed papers are
"Critical role of quantum dynamical effects in the Raman spectroscopy of liquid water"
Xinzijian Liu, Jian Liu*
Molecular Physics, 116(7-8), 755-779 (2018)
[Invited article for the special topic issue "Molecular Physics in China". Cover of the issue.]